
Registration Of Drug Product In Philippines (2025)


Overview
In the Philippines, the registration of a drug product is regulated by the Food and Drug Administration (FDA Philippines) under the Department of Health (DOH). The registration process requires submission of a comprehensive dossier in the ASEAN Common Technical Dossier (ACTD) format, organized into four main parts (Parts I to IV), covering administrative, quality, safety, and efficacy information of the drug product.
Pharmaceutical companies seeking to market a drug in the Philippines must submit a complete dossier that includes detailed information on the product’s quality, safety, efficacy, manufacturing process, and labeling. The process begins with the submission of a formal application through online via mail, which is first reviewed for completeness before undergoing technical evaluation by the Center for Drug Regulation and Research (CDRR). This evaluation assesses the scientific data, clinical evidence, and compliance with Philippine regulatory standards and Good Manufacturing Practices (GMP).
For imported products, a Certificate of Pharmaceutical Product (CPP) or equivalent document confirming marketing authorization in the country of origin or a recognized reference country is typically required. The approval timeline varies depending on the classification of the drug (e.g., new drug, generic, biologic), with innovative or new chemical entities (NCEs) generally requiring a more detailed and lengthy review.
Once approved, the product is granted a Certificate of Product Registration (CPR), typically valid for 3 to 5 years, depending on the product type. Upon expiry, renewal is necessary to maintain market authorization. Post-registration, companies are obligated to comply with pharmacovigilance and reporting requirements, ensuring continued monitoring of product safety and efficacy within the Philippine market.
Registration Process
Registration Dossier Structure
Structure is aligned with ASEAN Common Technical Dossier (ACTD) and local regulatory requirements (CDRR, FDA Circulars.
Part I – Administrative Information
Cover letter, application form
Legalized documents (CPP, GMP, LOA) from country of origin
Company, MAH & manufacturer details
Part II – Quality Documentation
CTD Modules 2 & 3
API/Finished product specs, CoA, validation & stability data
Packaging and labeling details
Part III – Non-Clinical Data
Pharmacology and toxicology summaries
Non-clinical study reports (if required)
Justification for omission (if based on literature or waiver)
Part IV – Clinical Data
Clinical trial summaries or full reports
Bioequivalence (BE) study reports (mandatory for generics unless a biowaiver is justified)
Literature data for well-established use or bibliographic submission
Risk-benefit summary
|
_
_
_
_
_
_
Fill up the form correctly and send an email to pair@fda.gov.ph for DTN
DTN (Document tracking number) receive with application
Payment
Dowload application form (IAF) from www.fda.gov.ph
Registration Fees
Pre-assessment
Submission
|
Fee Payment
Evaluation
Issuance of Certificate of Product Registration
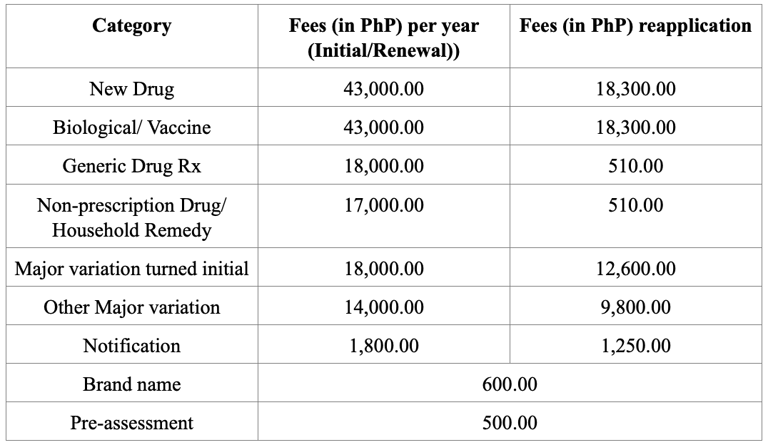
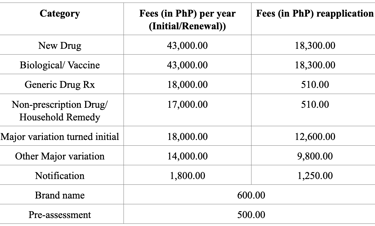
Note - All fees are subject to an additional PhP 10.00 or 1% of the application fee, whichever is higher, for Legal Research Fee (LRF).

